R course
Daniel Vaulot
2023-01-18
![]()
![]()
![]()
![]()
05 - Metabarcode processing with dada2
Data used
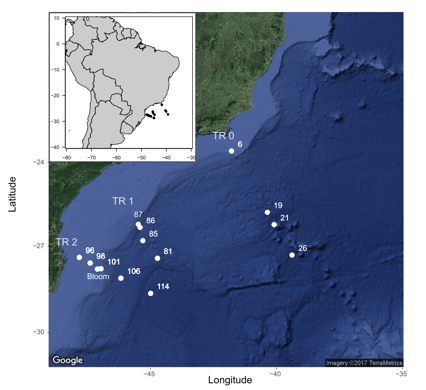
The samples originate from the CARBOM cruise (2013) off Brazil.
Samples have been sorted by flow cytometry and 3 genes have been PCR amplified :
- 18S rRNA - V4 region
- 16S rNA with plastid
- nifH
The PCR products have been sequenced by 1 run of Illumina 2*250 bp. The data consist of the picoplankton samples from one transect and fastq files have been subsampled with 1000 sequences per sample.
References
- Gerikas Ribeiro C, Marie D, Lopes dos Santos A, Pereira Brandini F, Vaulot D. (2016). Estimating microbial populations by flow cytometry: Comparison between instruments. Limnol Oceanogr Methods 14:750–758.
- Gerikas Ribeiro C, Lopes dos Santos A, Marie D, Brandini P, Vaulot D. (2018). Relationships between photosynthetic eukaryotes and nitrogen-fixing cyanobacteria off Brazil. ISME J in press.
- Gerikas Ribeiro C, Lopes dos Santos A, Marie D, Helena Pellizari V, Pereira Brandini F, Vaulot D. (2016). Pico and nanoplankton abundance and carbon stocks along the Brazilian Bight. PeerJ 4:e2587.
Compute number of paired reads
# create an empty data frame
df <- data.frame()
# loop through all the R1 files (no need to go through R2 which should be the same)
for(i in 1:length(fns_R1)) {
# use the dada2 function fastq.geometry
geom <- fastq.geometry(fns_R1[i])
# extract the information on number of sequences and file name
df_one_row <- data.frame (n_seq=geom[1], file_name=basename(fns_R1[i]) )
# add one line to data frame
df <- bind_rows(df, df_one_row)
} Display results
# display number of sequences
DT::datatable(df)
# plot the histogram with number of sequences
g <- ggplot(df, aes(x=n_seq)) +
geom_histogram( alpha = 0.5, position="identity", binwidth = 10) +
xlim(0, 2000)
print(g)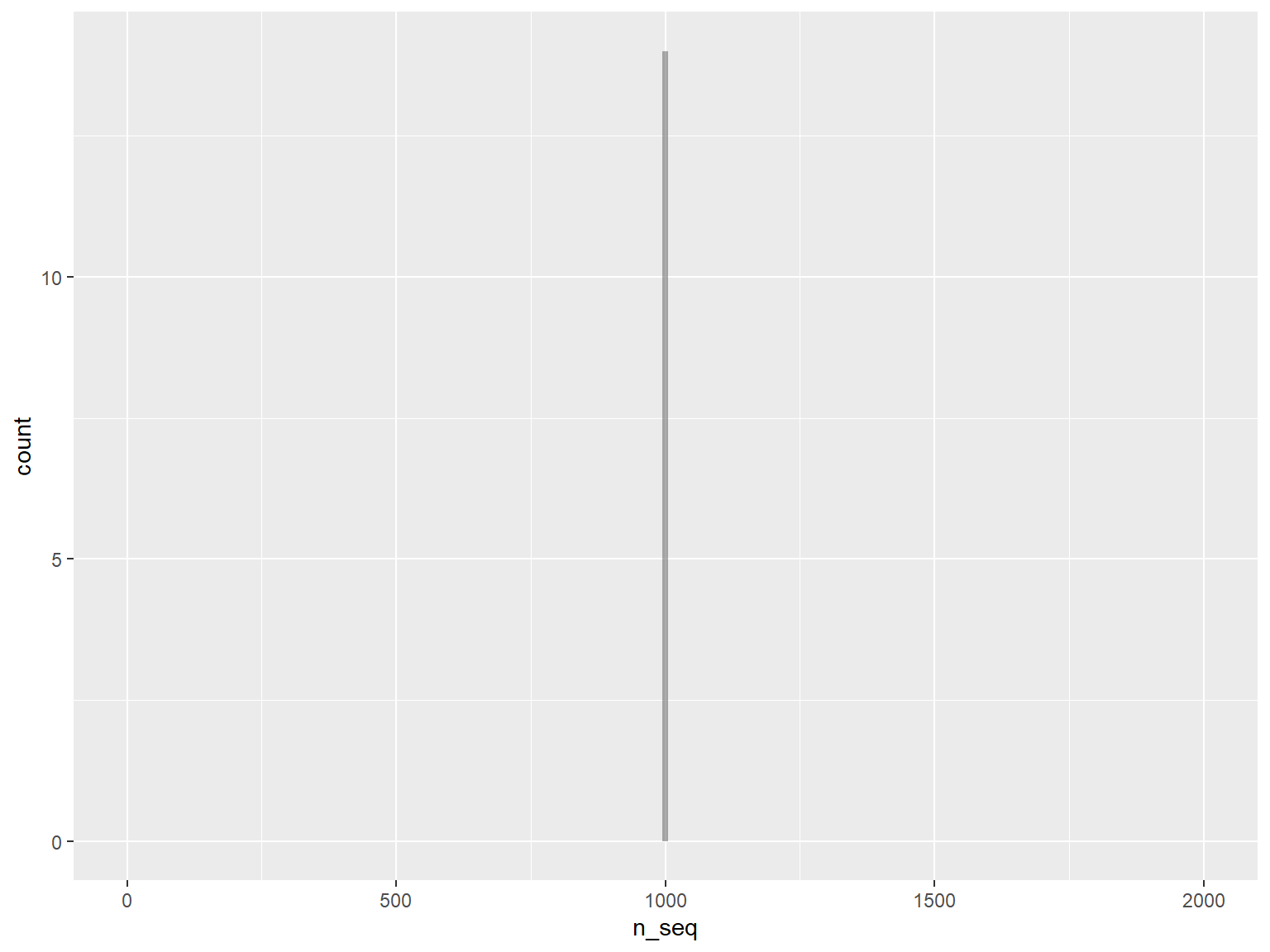
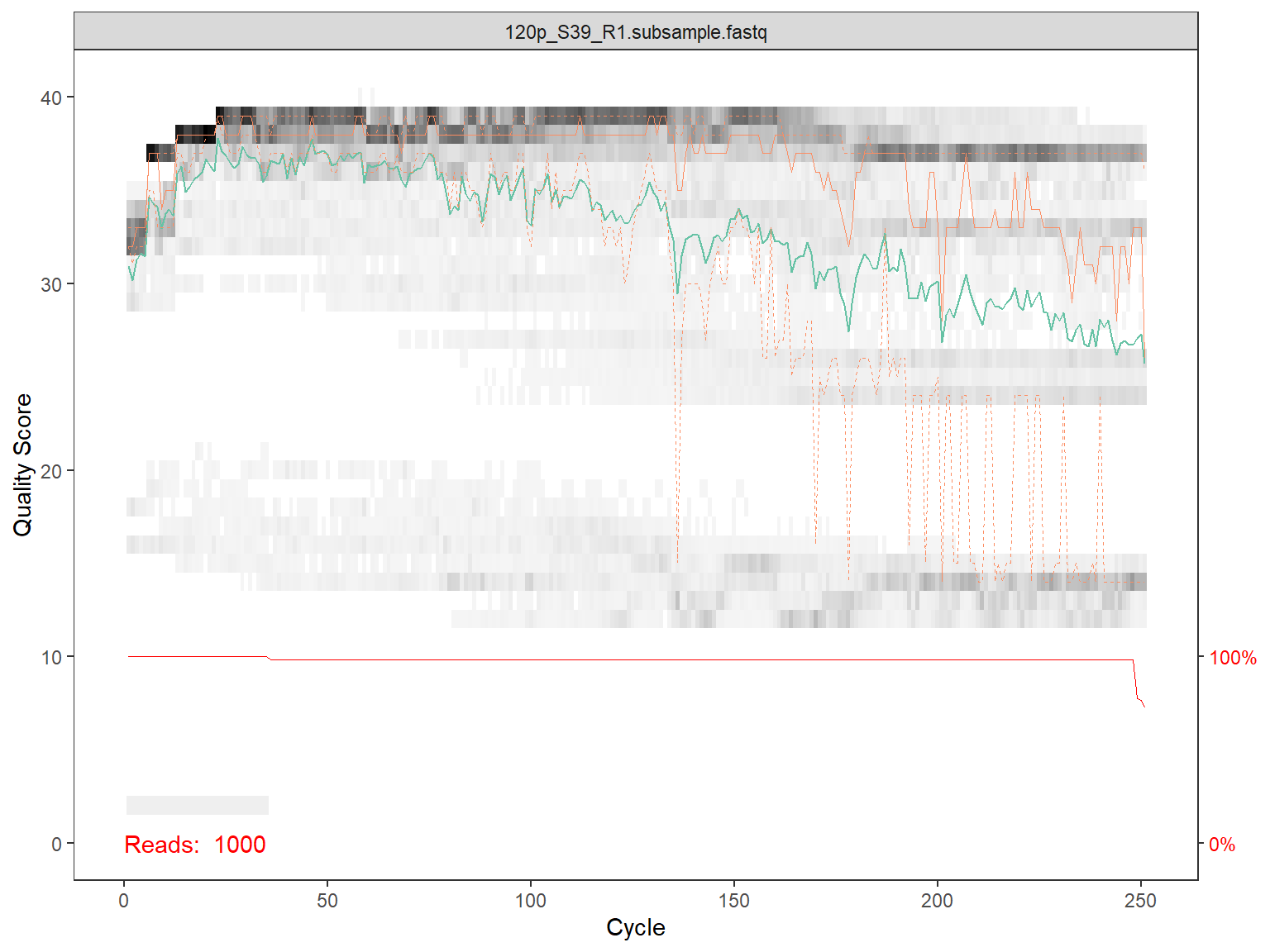
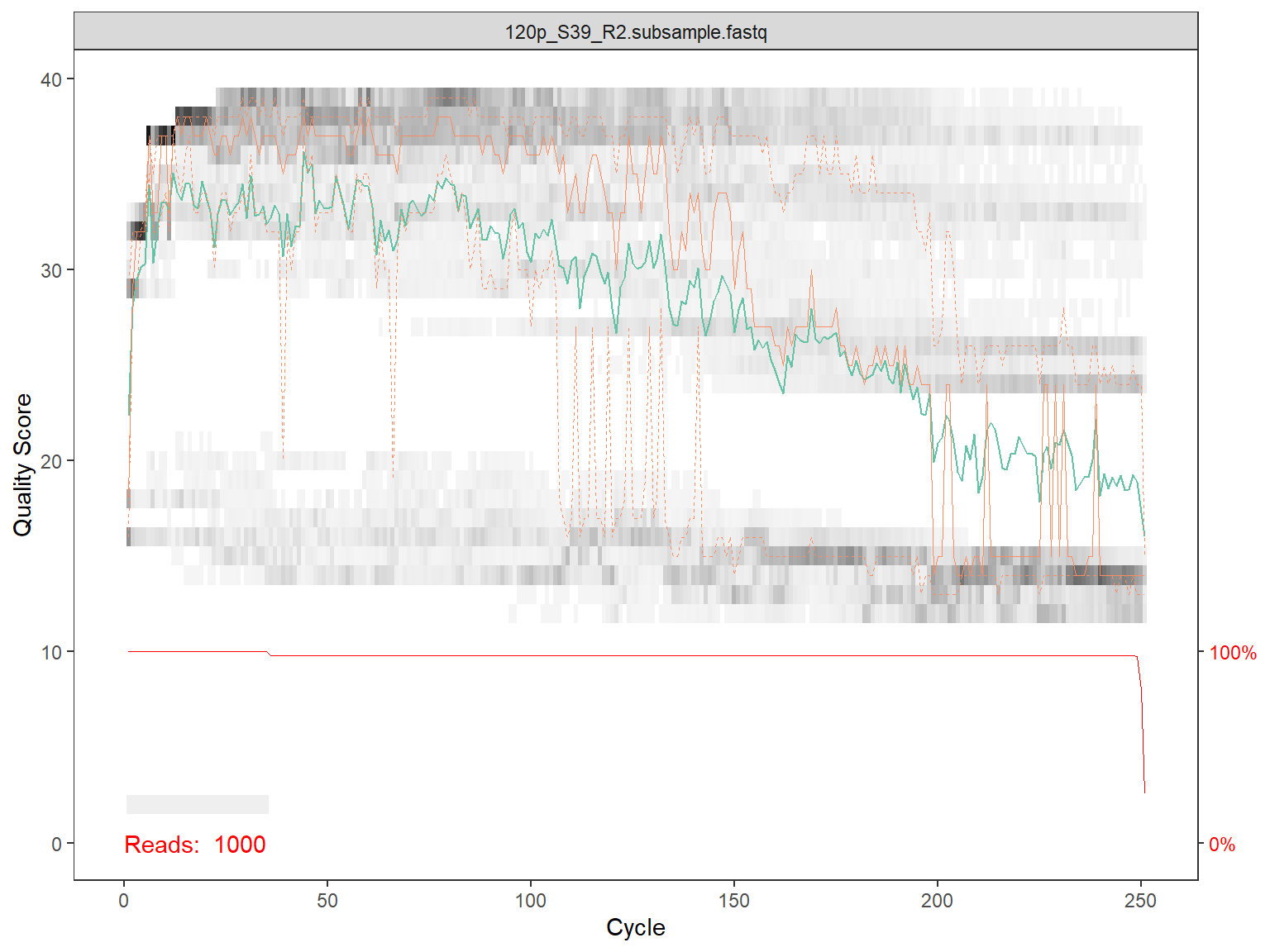
Plot quality for reads
for(i in 1:length(fns)) {
# Use dada2 function to plot quality
p1 <- plotQualityProfile(fns[i])
# Only plot on screen for first 2 files
if (i <= 2) {print(p1)}
# save the file as a pdf file (uncomment to execute)
p1_file <- paste0(qual_dir, basename(fns[i]),".qual.pdf")
ggsave( plot=p1, filename= p1_file,
device = "pdf", width = 15, height = 15, scale=1, units="cm")
} 
Learn error rates
The error rates are plotted.
R1
err_R1 <- learnErrors(filt_R1, multithread=FALSE)
plotErrors(err_R1, nominalQ=TRUE)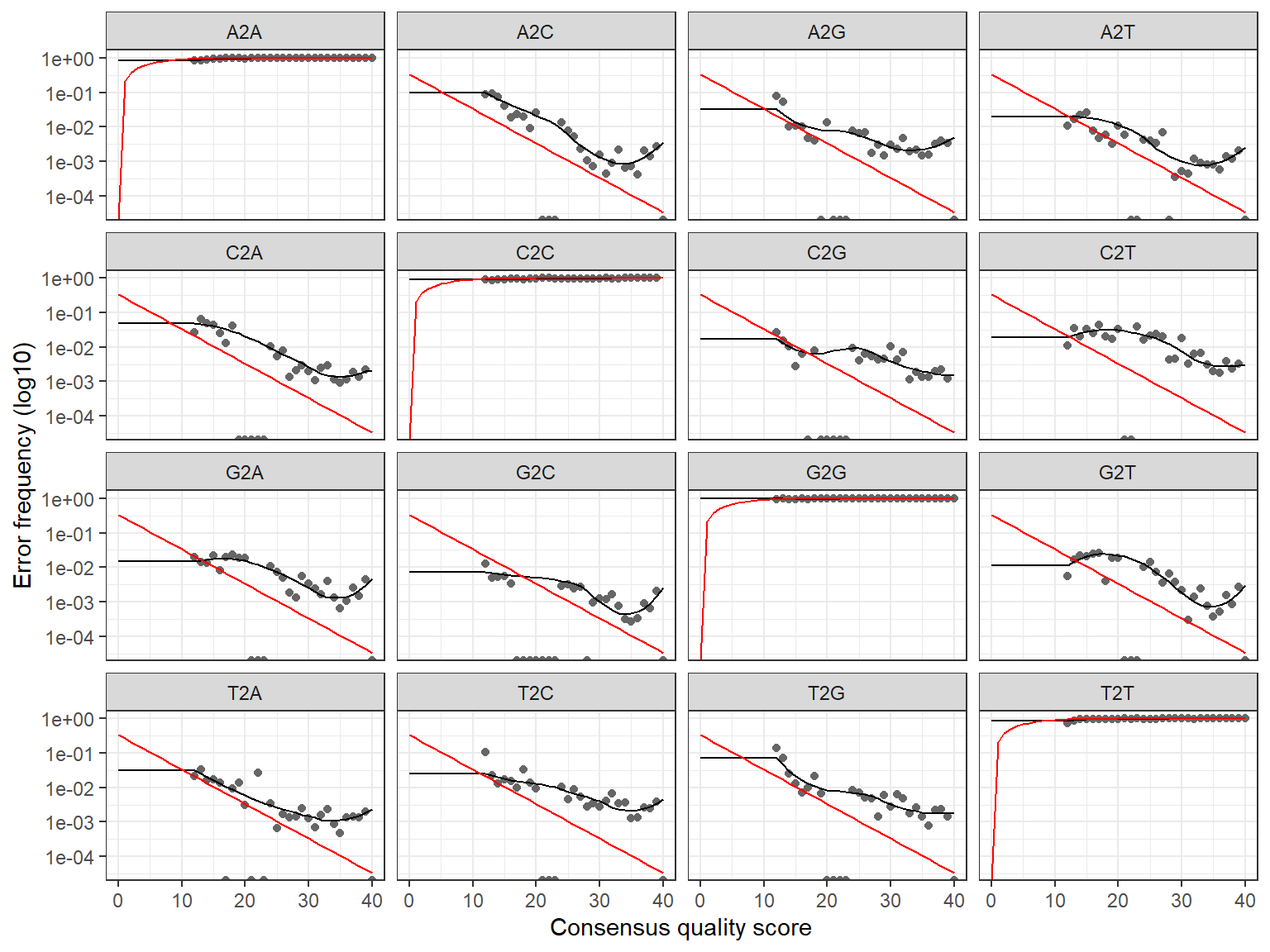
1581480 total bases in 6876 reads from 14 samples will be used for learning the error rates.R2
err_R2 <- learnErrors(filt_R2, multithread=FALSE)
plotErrors(err_R2, nominalQ=TRUE)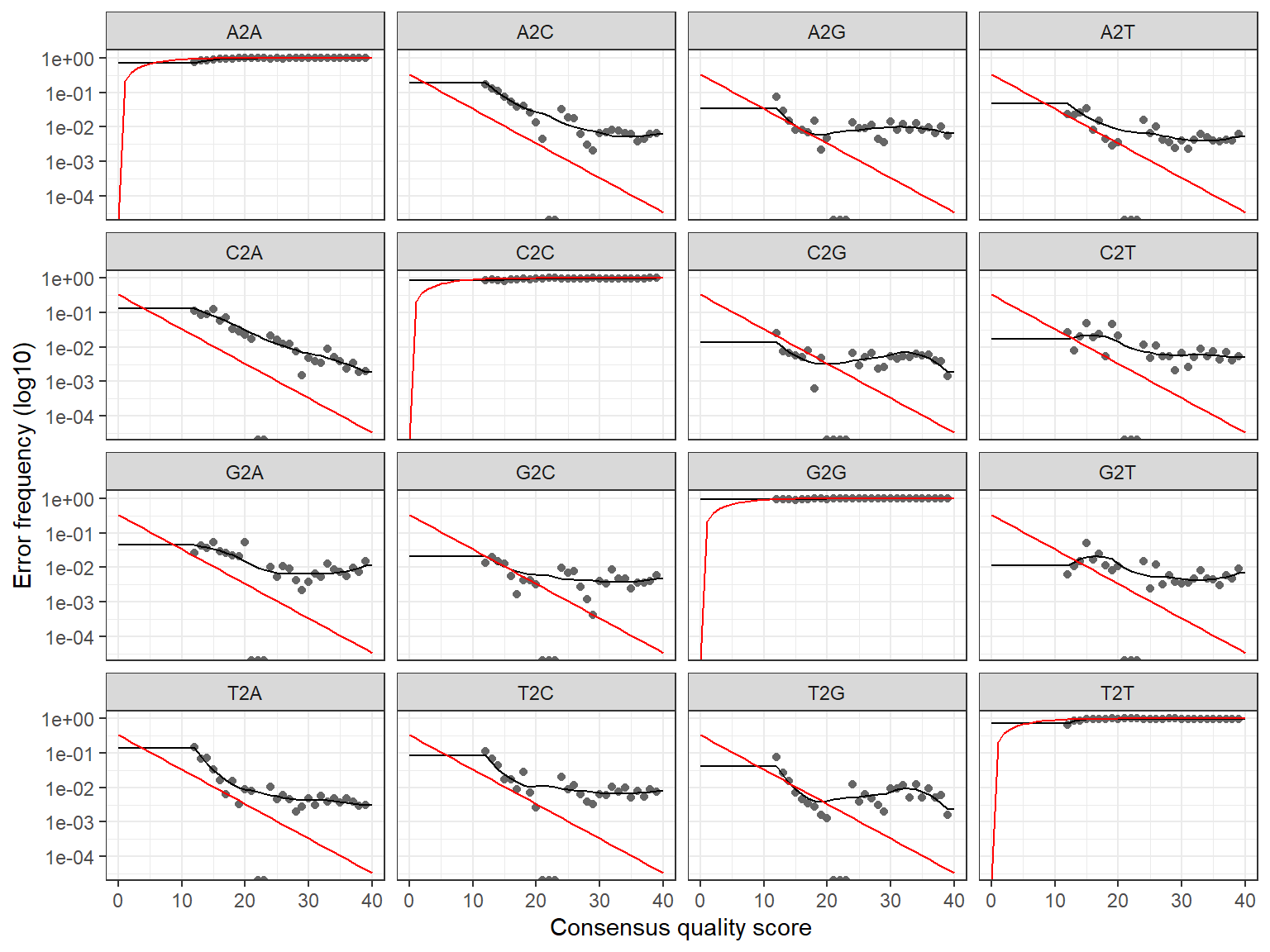
1505844 total bases in 6876 reads from 14 samples will be used for learning the error rates.