R course
Daniel Vaulot
2023-01-26
![]()
![]()
![]()
![]()
Metabarcode analysis - Phyloseq
Phyloseq R library
![]()
See in particular tutorials for
Get ASVs, read abundance and metadata all together
Filter and regroup the data
Bar plots, Alpha and Beta diversity computations
Data
This tutorial uses a reduced metabarcoding dataset obtained by C. Ribeiro and A. Lopes dos Santos. This dataset originates from the CARBOM cruise in 2013 off Brazil and corresponds to the 18S V4 region amplified on flow cytometry sorted samples (see pptx file for details) and sequenced on an Illumina run 2*250 bp analyzed with mothur.
Reference
- Gérikas Ribeiro, C., Dos Santos, A.L., Marie, D., Brandini, F.P. & Vaulot, D. 2018. Small eukaryotic phytoplankton communities in tropical waters off Brazil are dominated by symbioses between Haptophyta and nitrogen-fixing cyanobacteria. ISME Journal. 12:1360–74.
OTU
- rows = OTUs or ASVs
- cols = samples
- cells = number of reads

Table OTU - OTU abundance
Taxonomy
- rows = OTUs or ASVs
- cols = taxonomy ranks
- cells = taxon name
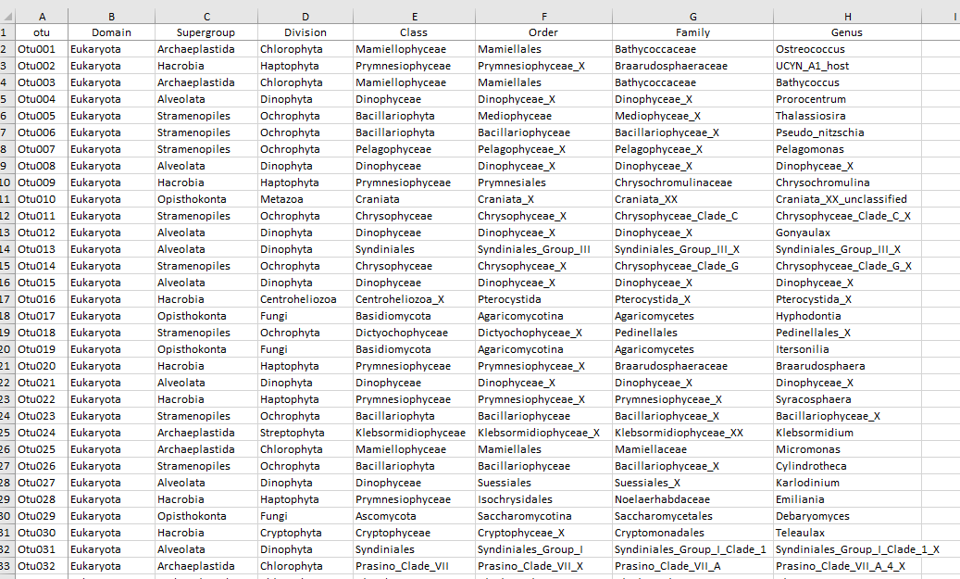
Table Taxo - OTU taxonomy
Samples
- rows = samples
- cols = metadata type
- cells = metadata values
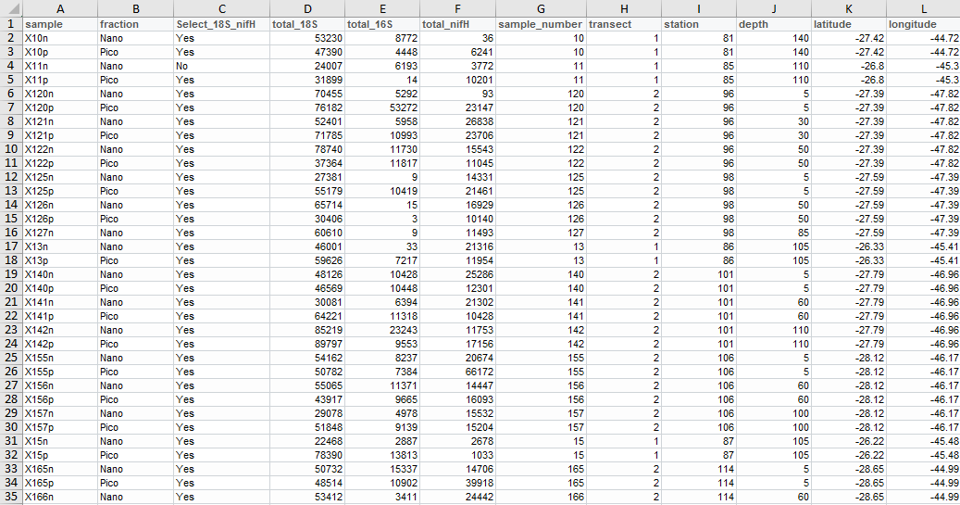
Table Samples
Basic bar graph based on Division
plot_bar(carbom, fill = "Division")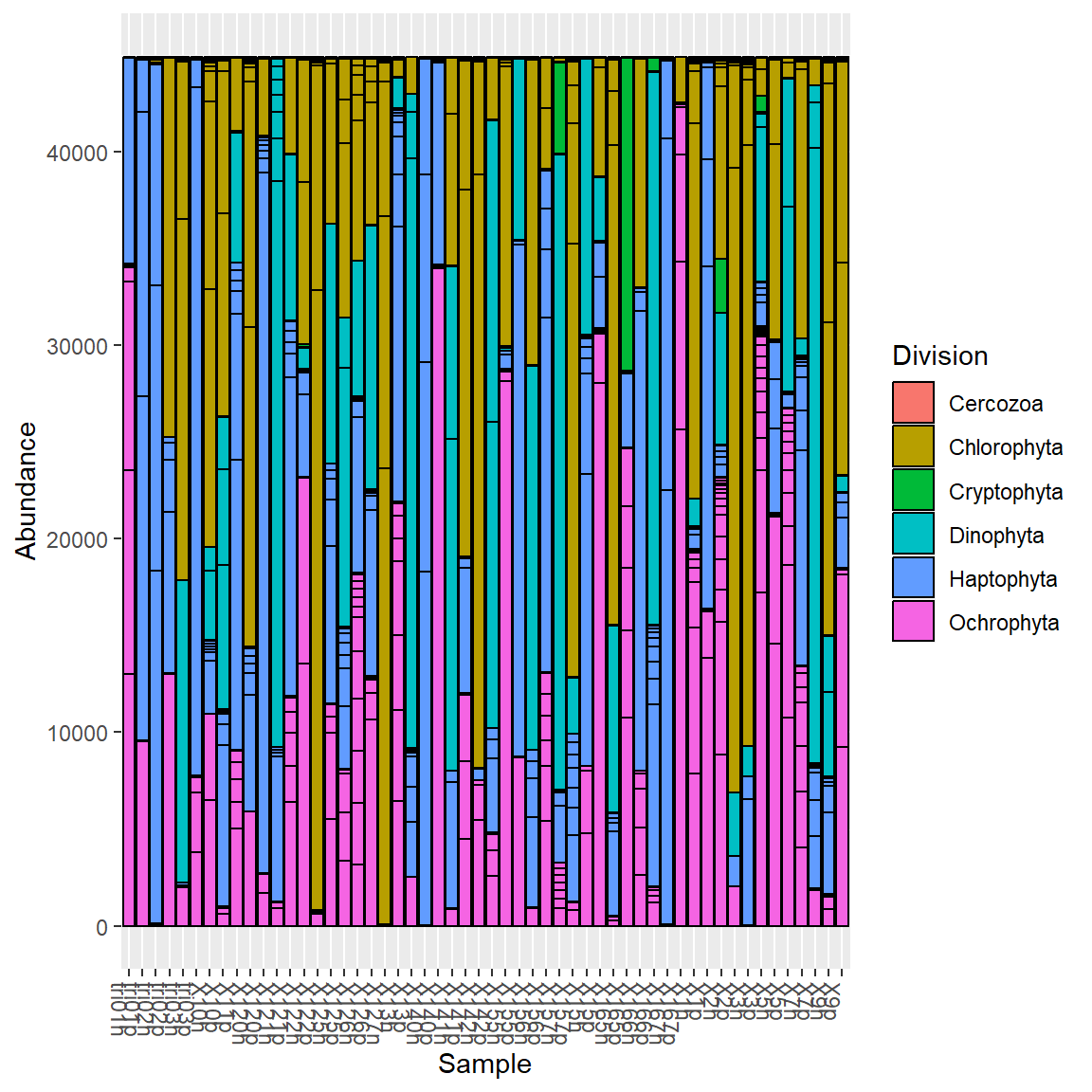
Remove OTUs boundaries.
# This is done by adding ggplot2 modifier.
plot_bar(carbom, fill = "Division") +
geom_bar(aes(color=Division,
fill=Division),
stat="identity",
position="stack")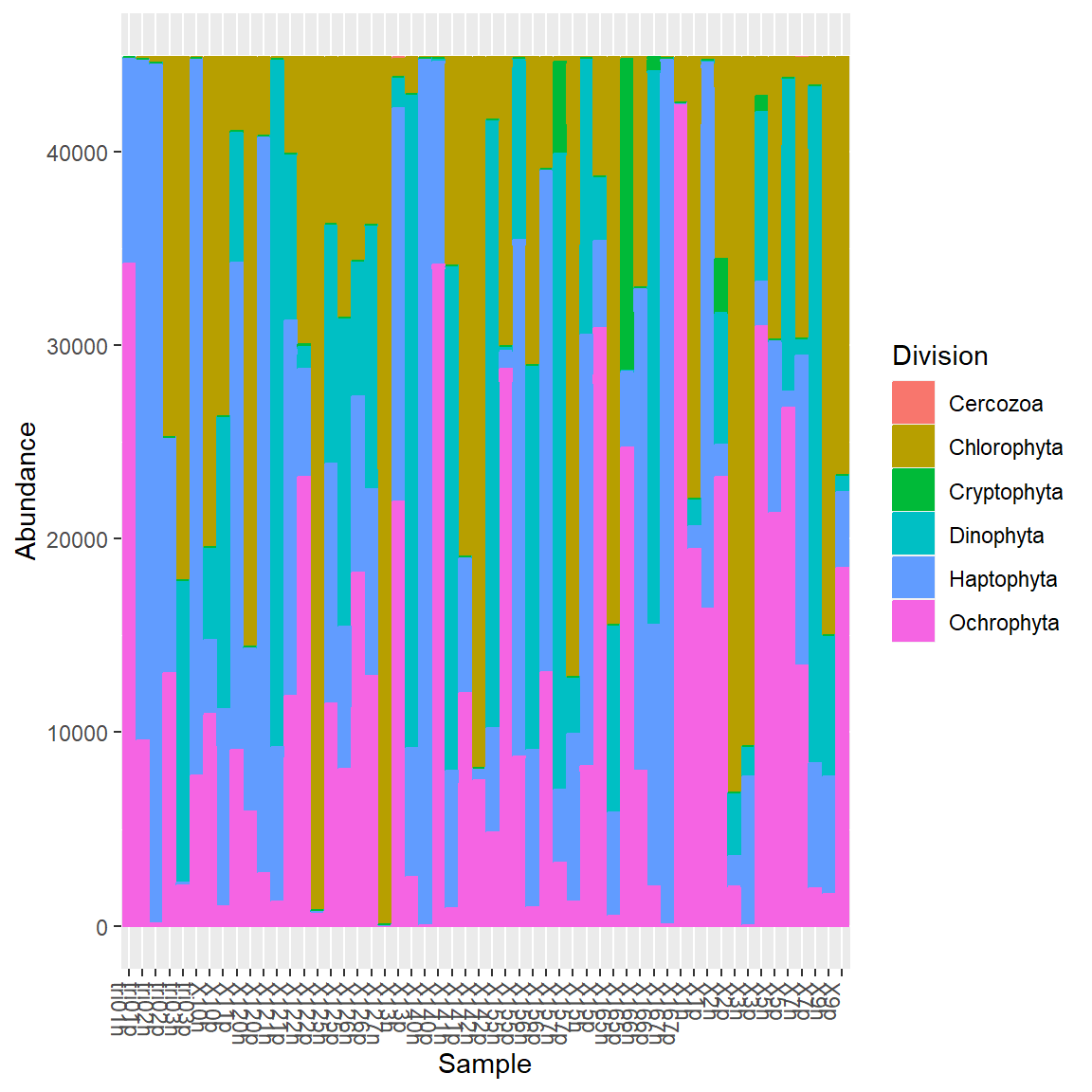
Group together Pico vs Nano samples.
carbom_fraction <- merge_samples(carbom, "fraction")
plot_bar(carbom_fraction, fill = "Division") +
geom_bar(aes(color=Division,
fill=Division),
stat="identity",
position="stack")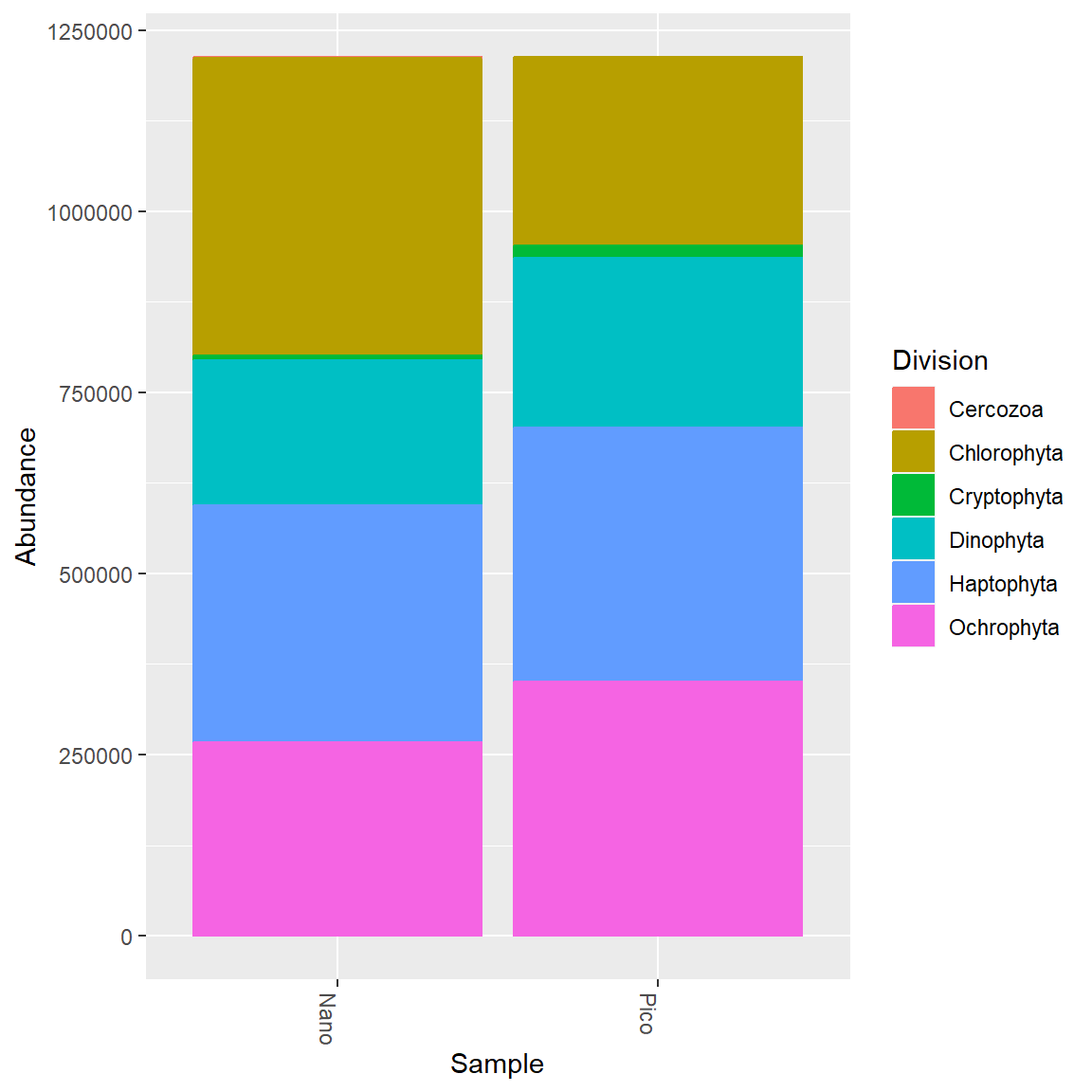
More grouping
# Keep only Chlorophyta
# Color according to genus.
# Pico vs Nano/Surface vs Deep.
carbom_chloro_ps <- subset_taxa(carbom,
Division %in% c("Chlorophyta"))
# Transform from phylseq to dataframe
carbom_chloro_df <- psmelt(carbom_chloro_ps) %>%
# Group by fraction and level
group_by(fraction, level) %>%
# Compute relative % for each group
mutate(Abundance_pct = Abundance/sum(Abundance) * 100)
# Use ggplot directly
ggplot(carbom_chloro_df) +
geom_bar(aes(x= Division, y = Abundance_pct, fill=Genus),
stat="identity",
position="stack") +
facet_grid(rows = vars(level), cols=vars(fraction))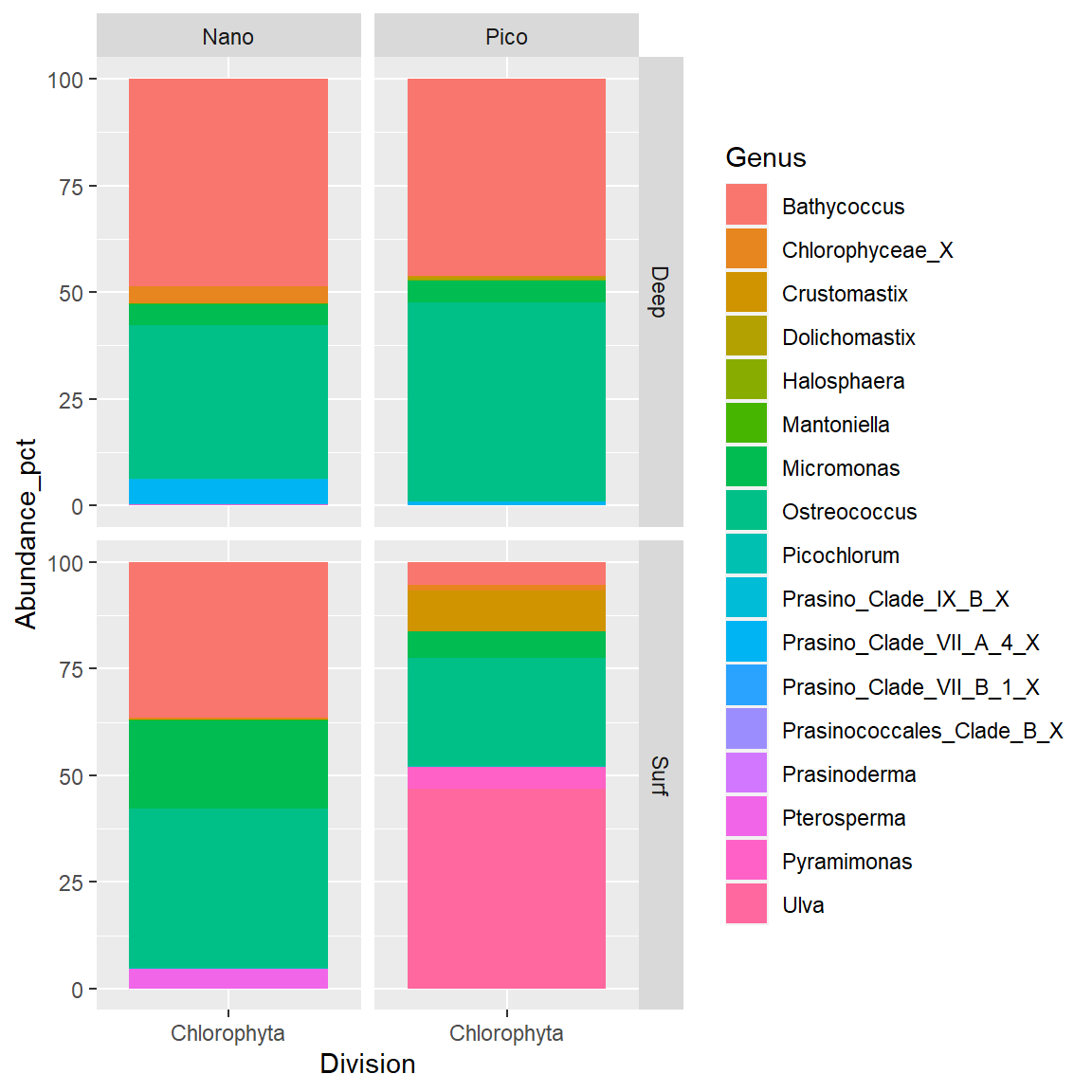
Diversity indexes
- Chao1: richness estimator
- Shannon: diversity estimator.
Other include: “Observed”, “ACE”, “Simpson”, “InvSimpson”, “Fisher”
plot_richness(carbom,
measures=c("Chao1", "Shannon"))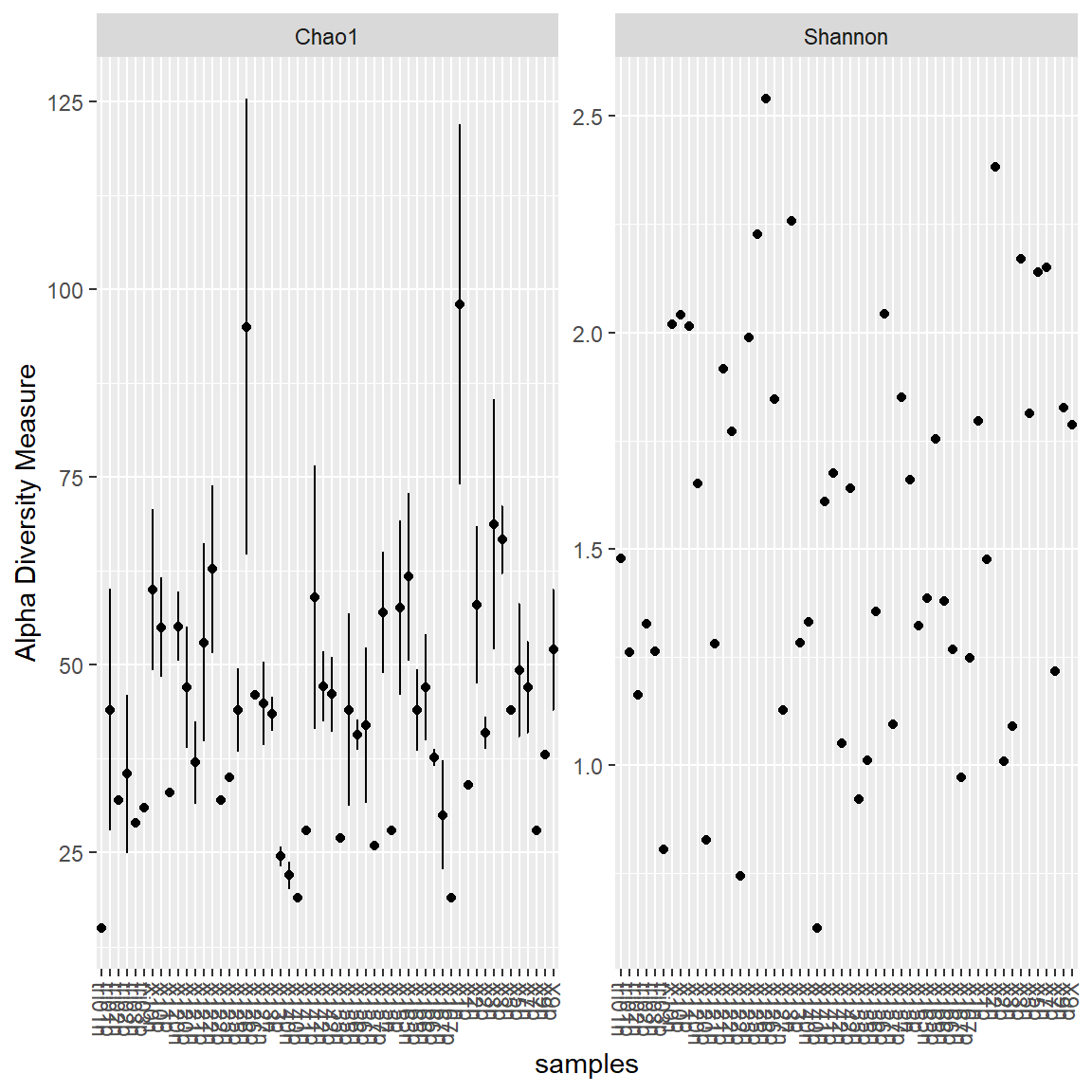
Regroup together samples by level and fraction.
plot_richness(carbom, measures=c("Chao1", "Shannon"),
x="level",
color="fraction")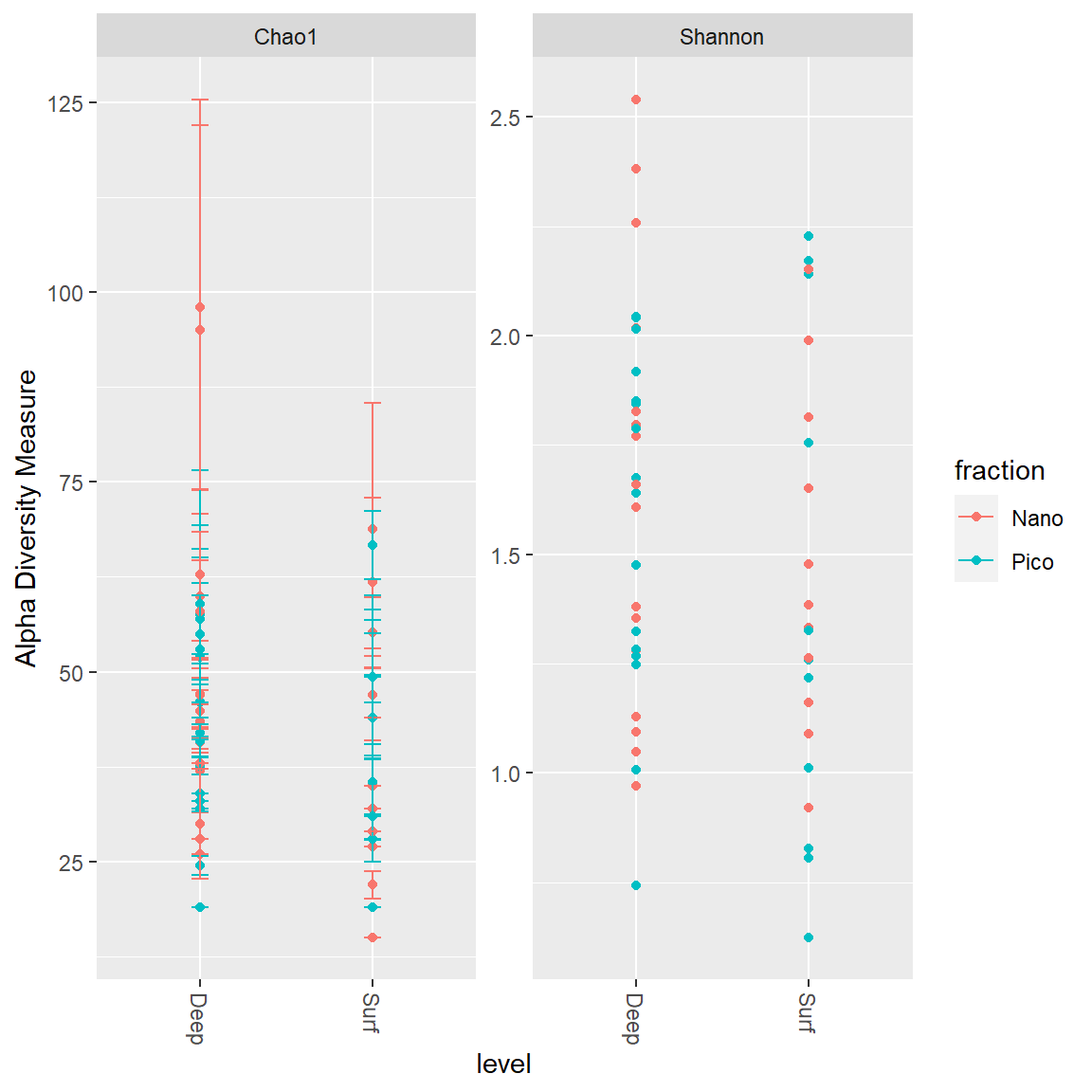
Plot OTUs
plot_ordination(carbom,
carbom.ord,
type="taxa",
color="Division",
title="OTUs")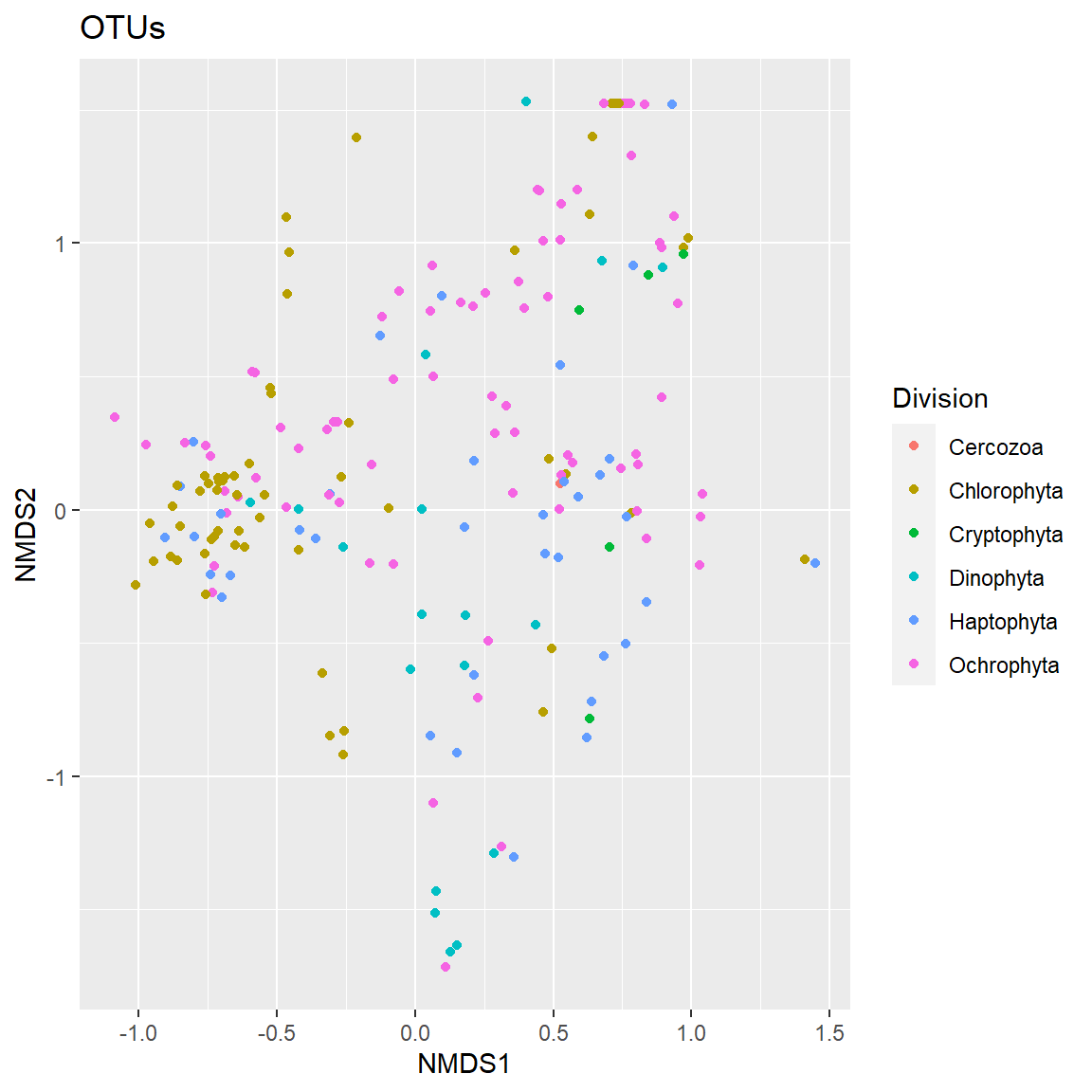
Plot OTUs
A bit confusing, so make it more easy to visualize by breaking according to taxonomic division.
plot_ordination(carbom,
carbom.ord,
type="taxa",
color="Class",
title="OTUs",
label="Class") +
facet_wrap(vars(Division), 3)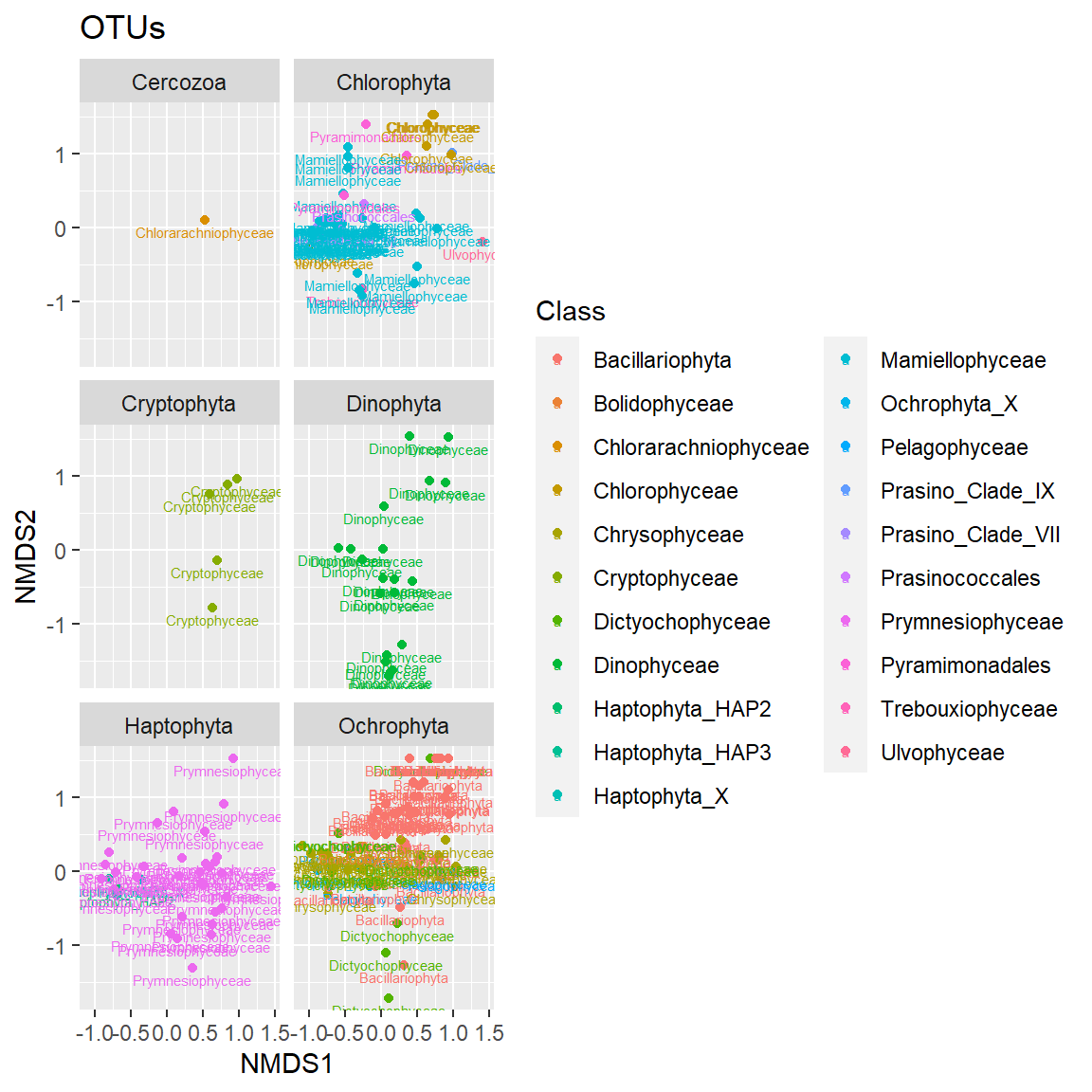
Plot Samples
Enlarge the points to make it more easy to read.
plot_ordination(carbom,
carbom.ord,
type="samples",
color="fraction",
shape="level",
title="Samples") +
geom_point(size=3)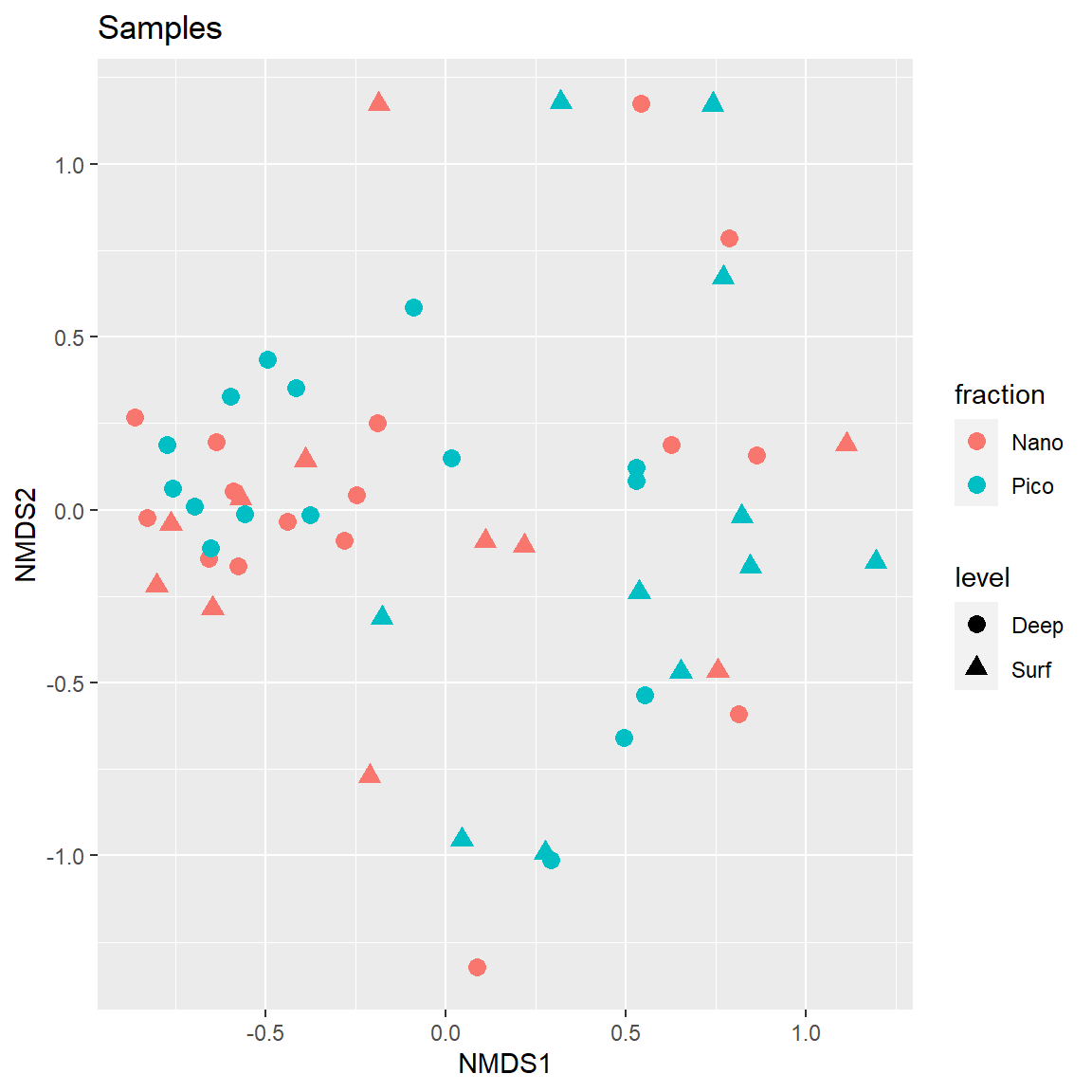
Plot both OTUs and Samples
Two different panels
plot_ordination(carbom,
carbom.ord,
type="split",
color="Division",
shape="level",
title="biplot",
label = "station") +
geom_point(size=3)
Basic heatmap using the default parameters.
plot_heatmap(carbom,
method = "NMDS",
distance = "bray")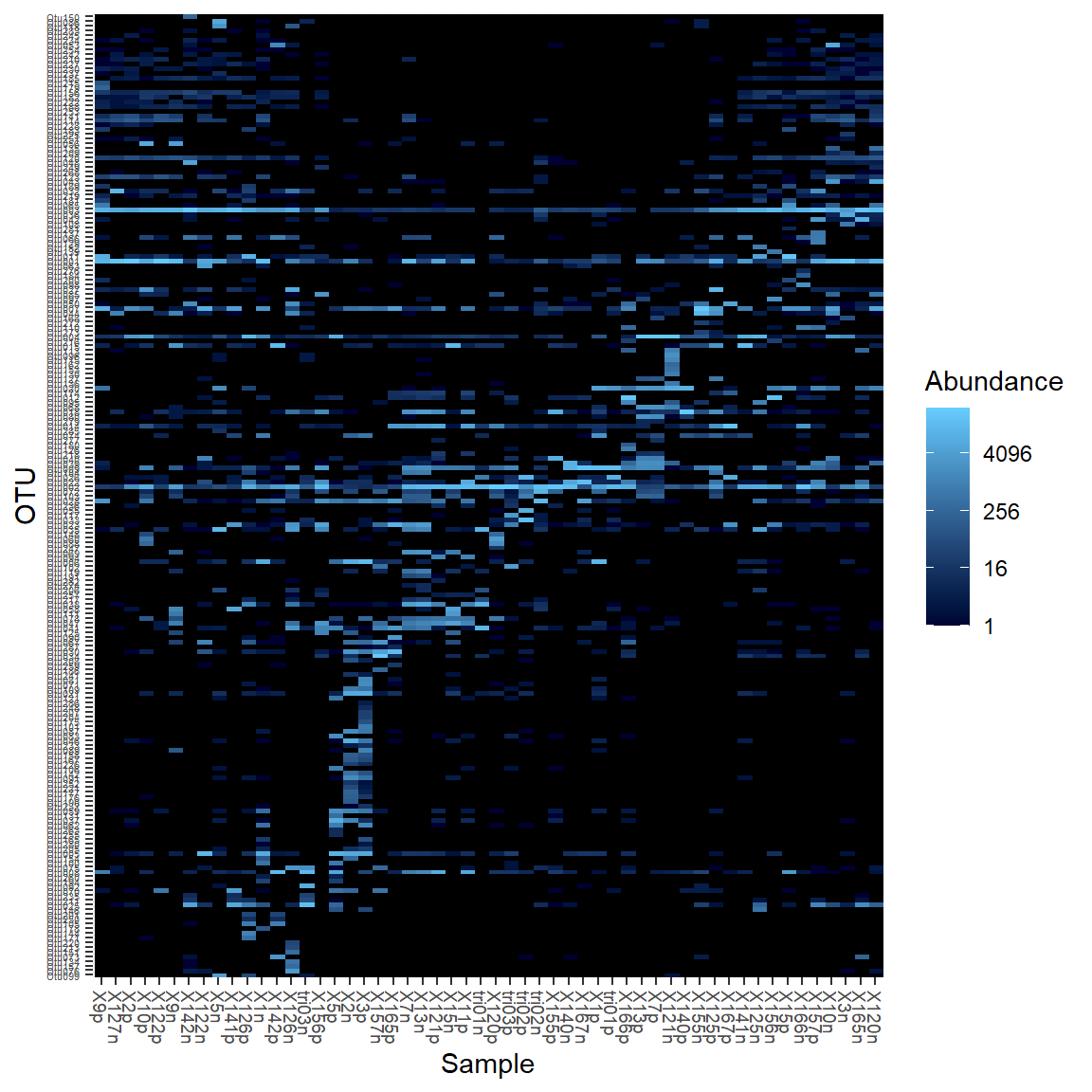
Consider only main OTUS
plot_heatmap(carbom_abund,
method = "NMDS",
distance = "bray")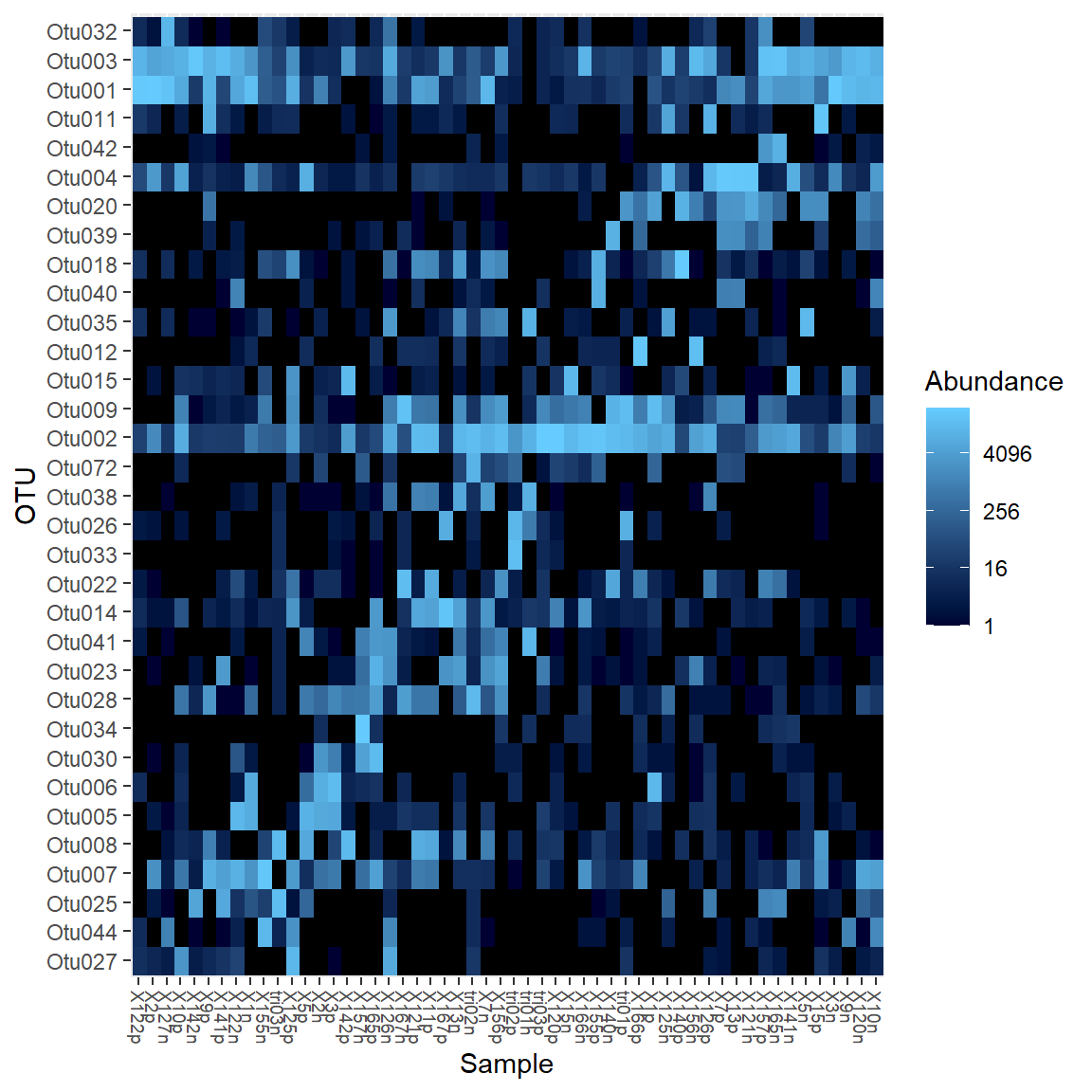
Change distances
It is possible to use different distances and different multivaraite methods. For example Jaccard distance and MDS and label OTUs with Class, order by Class. We can also change the Palette (the default palette is a bit ugly…).
plot_heatmap(carbom_abund,
method = "MDS",
distance = "(A+B-2*J)/(A+B-J)",
taxa.label = "Class",
taxa.order = "Class",
trans=NULL,
low="beige",
high="red",
na.value="beige")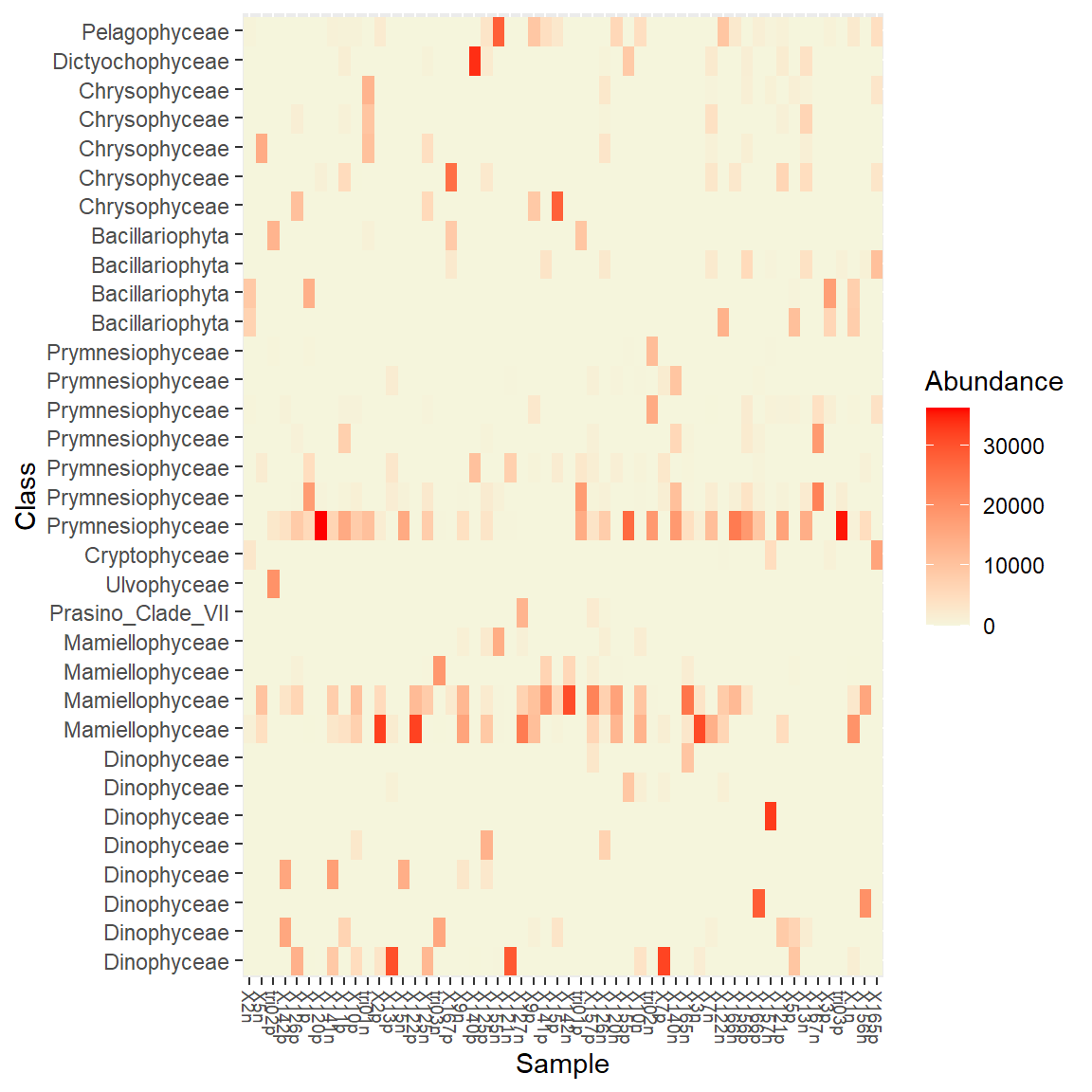
Heatmap for a specific taxonomy group.
For example we can taget the Chlorophyta and then label the OTUs using the Genus.
plot_heatmap(carbom_chloro_ps,
method = "NMDS",
distance = "bray",
taxa.label = "Genus",
taxa.order = "Genus",
low="beige",
high="red",
na.value="beige")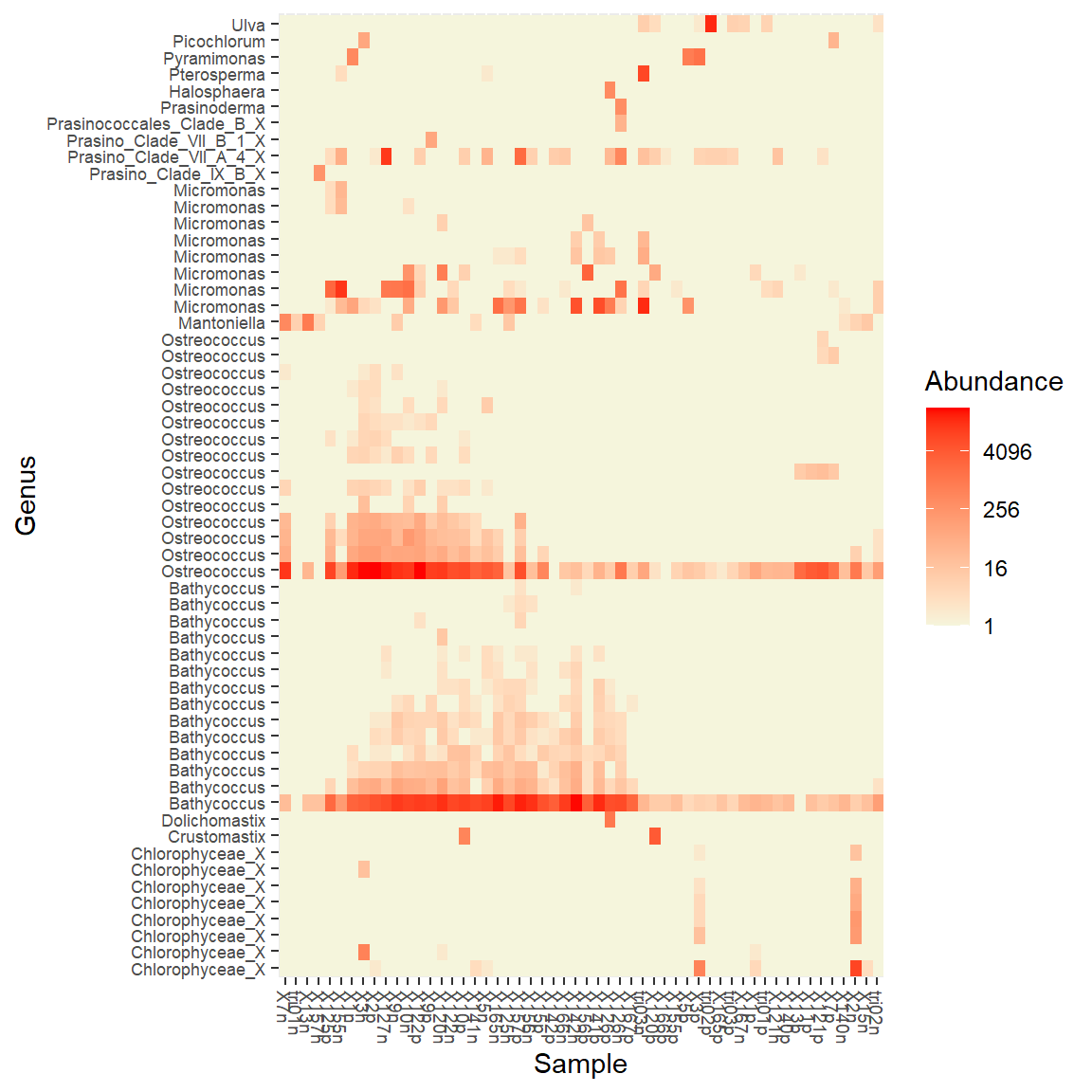
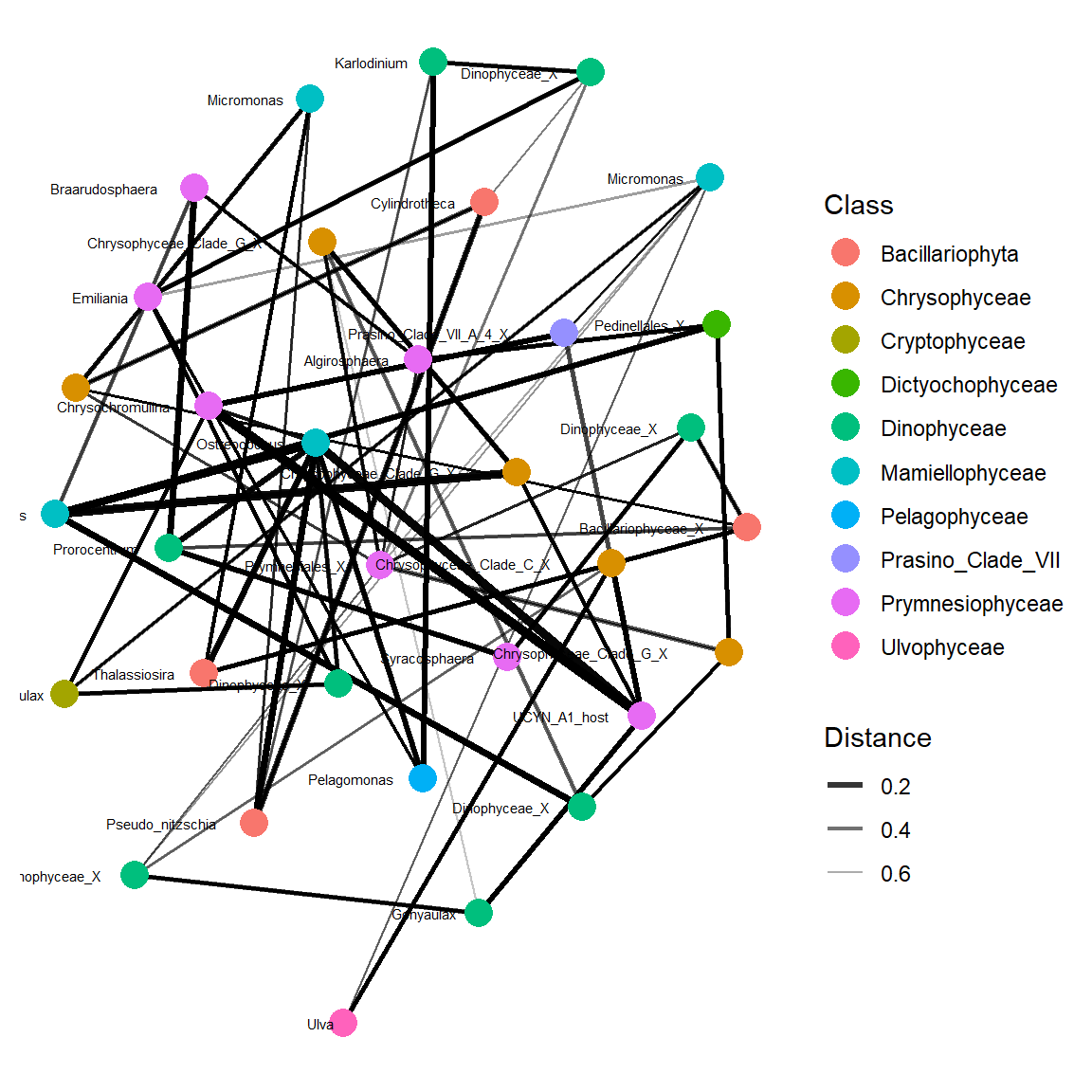
Simple network analysis
plot_net(carbom,
distance = "(A+B-2*J)/(A+B)",
type = "taxa",
maxdist = 0.7,
color="Class",
point_label="Genus")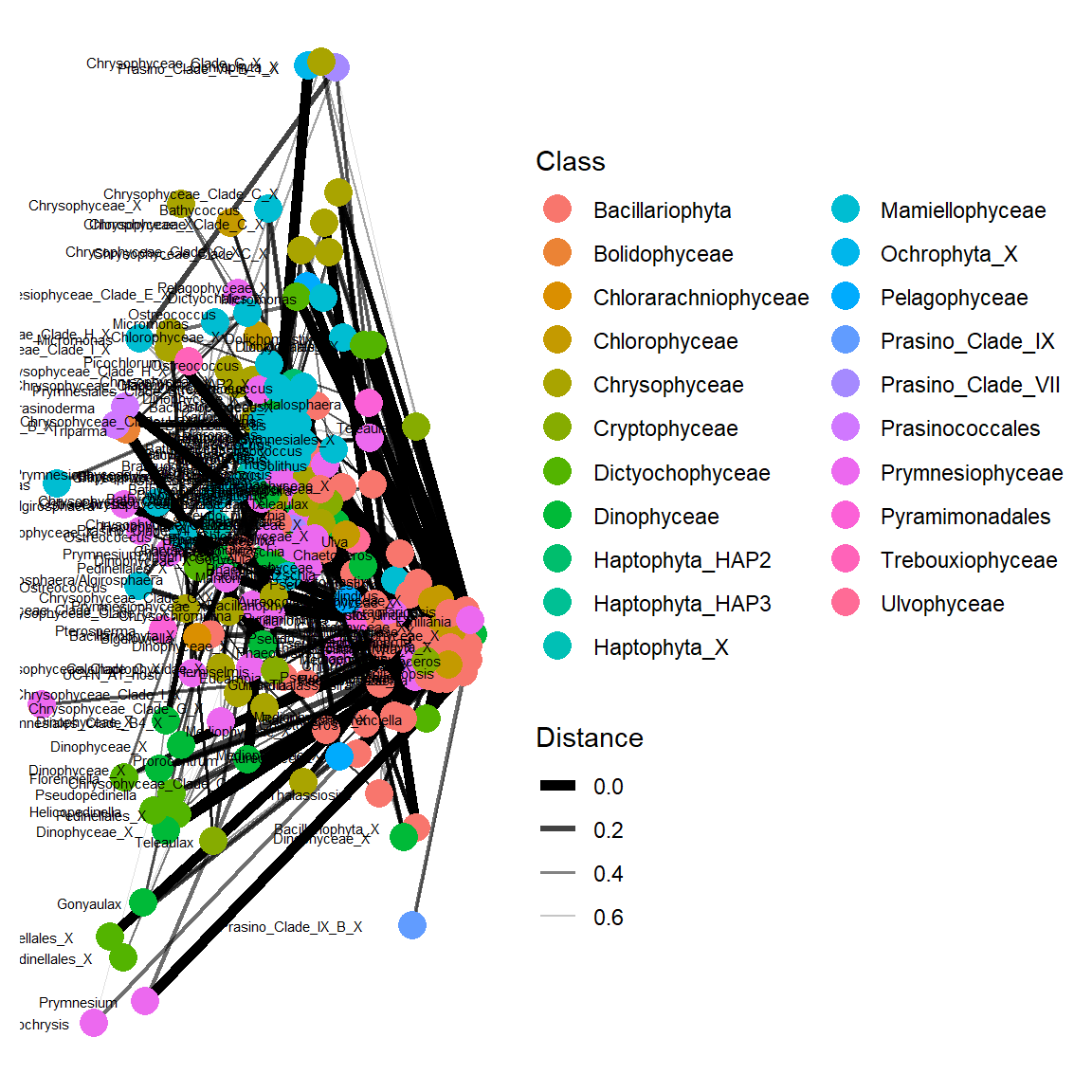
Simplify
Let us make it more simple by using only major OTUs
plot_net(carbom_abund,
distance = "(A+B-2*J)/(A+B)",
type = "taxa",
maxdist = 0.8,
color="Class",
point_label="Genus")